Le calcul des dérivées premières de la fonction "énergie" conduit au vecteur gradient

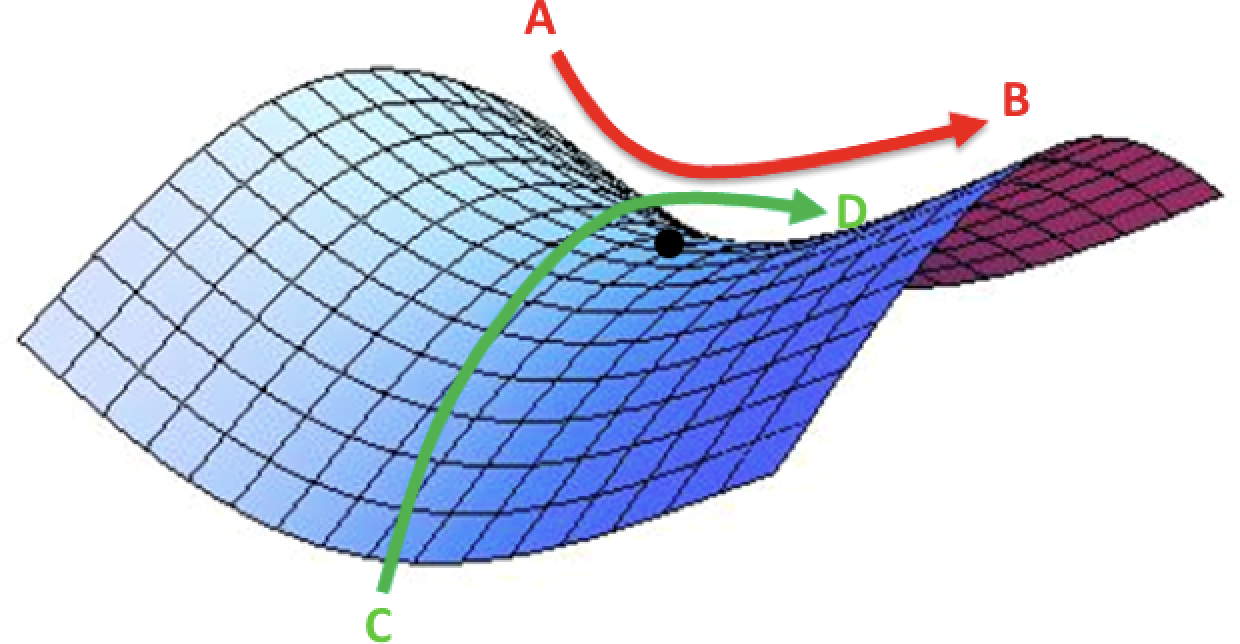
Quand ce vecteur est nul, on est sur un point "stationnaire" de l'espace des géométries, c'est la fin de l'optimisation. Il faut caractériser ce point stationnaire pour savoir si la géométrie de la molécule correspond à un minimum (puits) ou un état de transition (col ou saddle point). Cette caractérisation a besoin de la matrice des dérivées secondes, la matrice HESSIENNE de la fonction "énergie":

Pour ce faire on précise dans $CONTRL RUNTYP=HESSIAN au lieu de RUNTYP=ENERGY ou autre. Et on précise dans le groupe $FORCE la méthode de calcul: METHOD=ANALYTIC. Attention le calcul n'est rapide (analytique) que par la méthode Hartree-Fock. Pour les autres méthodes, comme la DFT par exemple, GAMESS procède par différentiation du gradient: il fait la differentiation numérique du gradient analytique METHOD=SEMINUM. Il peut aussi procéder par différentiation de l'énergie FULLNUM , selon les 3N coordonnées de la molécule ayant N atomes. Ce qui demande beaucoup de calculs si N est grand.
Le calcul du hessien permet de caractériser la géométrie comme un minimum ou un état de transition: l'analyse des modes de vibration montre en effet une unique fréquence imaginaire quand on a un état de transition. Le job 300235 montre un minimum dans le propene. Le job 300867 montre un état de transition dans le propene. La visualisation de la fréquence imaginaire (affichée comme négative) permet de mieux comprendre la différence entre ces 2 géométries. Le calcul indique une petite barrière de 9 kJ/mol.
Pour caractériser une géométrie lors d'une optimisation, on garde $CONTRL RUNTYP=OPTIMIZE. Le simple ajout du groupe $FORCE conduit au calcul du hessien et à son analyse vibrationnelle en fin d'optimisation.
Le calcul du hessien conduit à une analyse de ses valeurs propres dans le cadre de l'approximation harmonique. On simule ainsi le spectre IR des molécules. La précision est en général très bonne. A noter qu'au niveau Hartre-Fock on surestime un peu les valeurs des fréquences des modes normaux. Attention ChemCompute a tendance à reoptimiser la géométrie quand on la récupèred'un calcul précédent. Il faut décocher les cases correspondantes.
C'est à dire différentiation du gradient analytique. NVIB=2 indique de faire une double différenciation (un pas de chaque coté). NVIB=2 est obligatoire pour le calcul FULLNUM