Le calcul des dérivées premières de la fonction "énergie" conduit au vecteur gradient

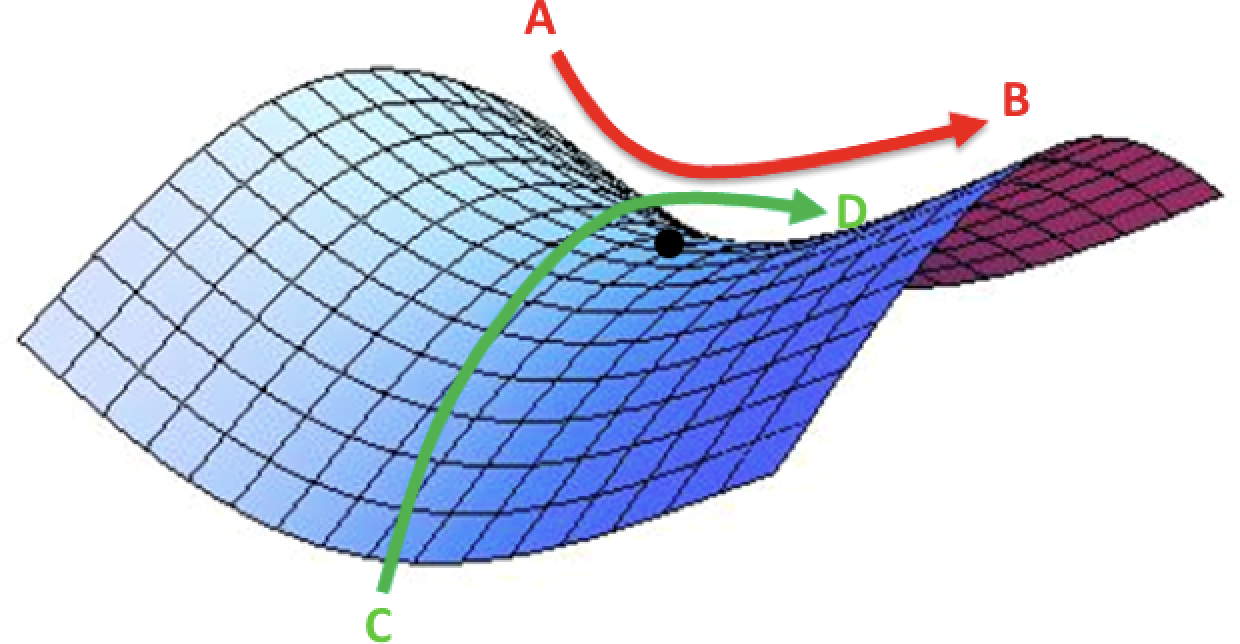
Quand ce vecteur est nul, on est sur un point "stationnaire" de l'espace des géométries, c'est la fin de l'optimisation. Il faut caractériser ce point stationnaire pour savoir si la géométrie de la molécule correspond à un minimum (puits) ou un état de transition (col ou saddle point). Cette caractérisation a besoin de la matrice des dérivées secondes, la matrice HESSIENNE de la fonction "énergie":

Pour ce faire on précise dans $CONTRL RUNTYP=HESSIAN au lieu de RUNTYP=ENERGY ou autre. Et on précise dans le groupe $FORCE la méthode de calcul: METHOD=ANALYTIC. Attention le calcul n'est analytique que si la symmétrie est C1 (la case "Use Symmetry" est décochée ou on met $CONTRL NOSYM=1 $END) - ou par la méthode Hartree-Fock qui peut gerer la symétrie.
Sinon, GAMESS procède par différentiation du gradient: il fait la differentiation numérique du gradient analytique METHOD=SEMINUM. Il peut aussi procéder par différentiation de l'énergie FULLNUM , selon les 3N coordonnées de la molécule ayant N atomes. Cette option demande beaucoup de calculs si N est grand.
Le calcul du hessien permet de caractériser la géométrie comme un minimum ou un état de transition: l'analyse des modes de vibration montre en effet une unique fréquence imaginaire quand on a un état de transition. Le job 300235 montre un minimum dans le propene. Le job 300867 montre un état de transition dans le propene. La visualisation de la fréquence imaginaire (affichée comme négative) permet de mieux comprendre la différence entre ces 2 géométries. Le calcul indique une petite barrière de 9 kJ/mol.
Pour caractériser une géométrie lors d'une optimisation, on garde $CONTRL RUNTYP=OPTIMIZE. Le simple ajout du groupe $FORCE conduit au calcul du hessien et à son analyse vibrationnelle en fin d'optimisation.
Le calcul du hessien conduit à une analyse de ses valeurs propres dans le cadre de l'approximation harmonique. On simule ainsi le spectre IR des molécules. La précision est en général très bonne. A noter qu'au niveau Hartre-Fock on surestime un peu les valeurs des fréquences des modes normaux.
L'interface de Chemcompute n'est pas parfaite. On peut devoir chercher les fréquences et leurs symétrie dans le fichier de sortie tout à la fin à "MODE FREQ".
Attention ChemCompute risque de reoptimiser la géométrie et d'ajouter des hydrogènes quand on la récupère d'un calcul précédent. Décocher les cases correspondantes permet de sécuriser votre travail.
C'est à dire différentiation du gradient analytique. NVIB=2 indique de faire une double différenciation (un pas de chaque coté). NVIB=2 est obligatoire pour le calcul FULLNUM
SUM ON I M(I) * (X(I,J)*X(I,K) + Y(I,J)*Y(I,K) + Z(I,J)*Z(I,K)) = DELTA(J,K)
MODE FREQ(CM**-1) SYMMETRY RED. MASS IR INTENS.
1 7.012 B3G 4.765358 0.000000
2 6.559 B2G 3.550460 0.000000
3 0.761 B1G 6.967054 0.000000
4 0.060 B1U 8.003475 0.000001
5 0.020 B2U 8.003963 0.000000
6 0.007 B3U 8.003921 0.000000
7 351.228 AU 2.287405 0.000000
8 432.878 B3U 7.124879 0.595580
9 608.482 AG 8.805001 0.000000
10 719.150 B3G 6.947709 0.000000
11 767.220 B1G 4.191303 0.000000
12 802.323 B3U 1.134185 0.758133
13 941.961 B2G 1.259581 0.000000